Voluntary Nationwide Recall of 0.9% Sodium Chloride for Injection USP 1000 mL in E3 Containers Due to the Potential for Particulate Matter and Leakage
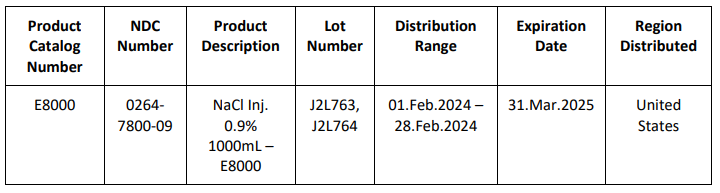
B. Braun Medical Inc. (B. Braun), is voluntarily recalling two (2) lots of 0.9% Sodium Chloride for Injection USP 1000mL in E3 containers within the United States to the consumer level. The voluntary recall has been initiated due to the potential for particulate matter and fluid leakage of the respective containers.
The affected batches were inadvertently released to the market prior to the completion of the required acceptance activities for embedded particulate matter which may result in leakage. To date, there have been no customer complaints received and there have been no reports of serious injury or death associated with this issue.
Risk Statement: There is a reasonable probability of embolic phenomena such as stroke or ischemia/infarct to other organs and possible infection if these particulates are not sterile that could lead to permanent damage or impairment of body function which could be life-threatening.
B. Braun has notified its distributors and customers by an official recall notice sent via certified registered mail and has arranged for the return of all recalled products. Facilities and distributors that have the product which is being recalled should discontinue use immediately and contact the B. Braun Medical Inc. Customer Support Department at 800-227-2862 Monday through Friday, 8 a.m. – 6 p.m. EST to arrange for product return.
Adverse reactions or quality problems experienced with this product, or questions about this recall may be reported to B. Braun’s Postmarket Surveillance Department by calling 1-833-425-1464.
Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail or by fax or call 1- 800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
This recall is being conducted with the knowledge of the U.S. Food and Drug
No members of BeneCard PBF were affected by this recall. Administration.