Earlier onset of atrial fibrillation (AF) is associated with increased risk of developing all-cause dementia, vascular dementia (VD), and Alzheimer disease (AD), according to a study published online Nov. 8 in JAMA Network Open.
Wenya Zhang, from the Chinese Academy of Medical Sciences & Peking Union Medical College in Beijing, and colleagues conducted a population-based cohort study using data from the U.K. Biobank to examine whether age at AF diagnosis is associated with risk of incident dementia and its subtypes. The main analysis included 433,746 participants.
The researchers found that compared to individuals without AF, the 30,601 with AF had increased risk of developing all-cause dementia and VD (adjusted hazard ratios, 1.42 and 2.06, respectively), but not AD. Younger age at AF onset was associated with increased risks of developing all-cause dementia, AD, and VD (adjusted hazard ratio per 10-year decrease, 1.23, 1.27, and 1.35, respectively). For developing all cause dementia, the highest hazard ratio was seen for individuals with AF diagnosed before age 65 years, followed by AF diagnosed at age 65 to 74 years (adjusted hazard ratios, 1.82 and 1.47, respectively) after propensity-score matching; the hazard ratio for AF diagnosed at ≥75 years was not significant. Results were similar for AD and VD.
“The quantitative manifestation of the association between AF onset age and incident dementia highlights the importance of monitoring cognitive function among AF patients, especially those younger than 65 years at diagnosis,” the authors write.
The Food and Drug Administration (FDA) is advising healthcare providers who administer the Moderna COVID-19 Vaccine (2023-2024 Formula) to individuals 6 months through 11 years of age to ensure that the correct volume of the vaccine (0.25 mL) is withdrawn from the vial, so that the correct dose is administered to the vaccine recipient.
The Moderna COVID-19 Vaccine (2023-2024 Formula) is authorized for use in individuals 6 months through 11 years of age.
Summary of the Issue
FDA has become aware that some healthcare providers may not recognize that the single dose vial of Moderna COVID-19 Vaccine (2023-2024 Formula) for use in individuals 6 months through 11 years of age contains notably more than 0.25 mL of the vaccine. Some healthcare providers may be withdrawing the entire contents of the vial to administer to an individual. However, the volume of a single dose of Moderna COVID-19 Vaccine (2023-2024 Formula) is only 0.25mL.
Information for Healthcare Providers
We are advising healthcare providers who administer the Moderna COVID-19 Vaccine (2023-2024 Formula) to individuals 6 months through 11 years of age to ensure that the correct volume of the vaccine is withdrawn from the vial, so that the correct dose is administered to the vaccine recipient. To provide clarification, the Dosage and Administration section of the Fact Sheet for Healthcare Providers Administering Vaccine has been revised to further clarify that 0.25mL should be withdrawn from the vial and that the vial and any excess volume should then be discarded.
Healthcare providers, parents and caregivers who have questions may contact FDA’s Center for Biologics Evaluation and Research (CBER) at ocod@fda.hhs.gov.
FDA Approves Agamree (vamorolone) for the Treatment of Duchenne Muscular Dystrophy
Catalyst Pharmaceuticals, Inc. has reported that Santhera Pharmaceuticals has obtained U.S. Food and Drug Administration (“FDA”) approval for Agamree® (vamorolone) oral suspension 40 mg/mL for use in treating Duchenne Muscular Dystrophy (DMD) in patients aged two years and older.
Agamree offers a novel corticosteroid treatment option for DMD, addressing a significant unmet medical need.
Agamree was granted Orphan Drug and Rare Pediatric Disease designations status for DMD in the U.S. and will be eligible for seven years of orphan drug exclusivity upon approval date and has issued pending patents that could provide protection until 2040.
In July 2023, Catalyst secured the exclusive North American license and commercial rights for Agamree from Santhera for DMD and other potential indications, bolstering its neuroscience commercial portfolio with a highly synergistic neuromuscular asset. As part of that transaction, Santhera will promptly transfer the approved New Drug Application for Agamree to Catalyst. The company plans to launch the product in Q1 2024.
Agamree’s unique mode of action is based on differential effects on glucocorticoid and mineralocorticoid receptors and modifying further downstream activity and, as such, is considered a novel corticosteroid with dissociative properties in maintaining efficacy, with a better-tolerated side effect profile. This mechanism of action may allow vamorolone to emerge as an effective alternative to the current standard of care corticosteroids in children, adolescents, and adult patients with DMD.
Exela Pharma Sciences, LLC, (Exela) is voluntarily recalling the products listed in the table below to the consumer level. Particulate matter identified as silicone was observed during routine inspection of retain samples.
Risk Statement: Administration of an injectable product that contains particulate matter may result in local irritation or swelling in response to the foreign material. If the particulate matter reaches the blood vessels it can travel to various organs and block blood vessels in the heart, lungs or brain which can cause stroke and even lead to death. Exela has not received any reports of adverse events related to this recall.
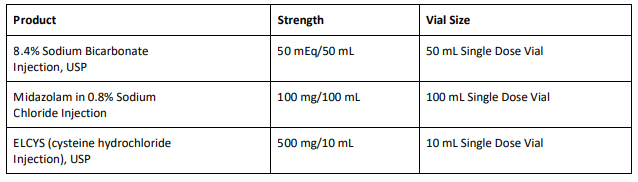
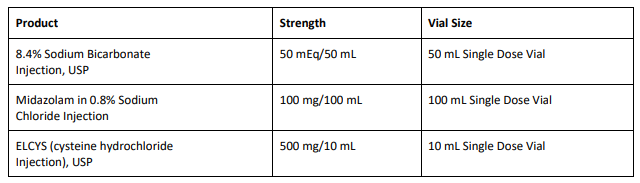
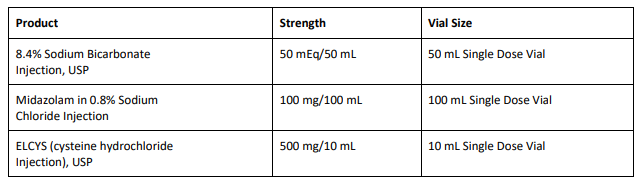
8.4% Sodium Bicarbonate Injection USP is used for treatment of metabolic acidosis and is packaged in a 50 mL glass single dose vials, 20 vials per carton Exela brand (Carton NDC: 51754-5001-5; Vial NDC: 51754- 5001-1, Figure 1) and 25 vials per carton Exela brand (Carton NDC: 51754-5001-4; Vial NDC: 51754-5001-1) and Civica brand (Carton NDC: 72572-740-20; Vial NDC: 72572-740-01, Figure 2).
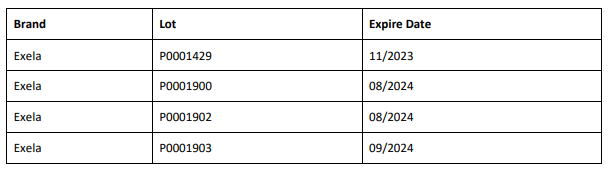
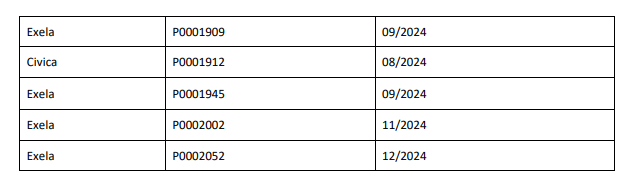
The affected 8.4% Sodium Bicarbonate Injection, USP, 50 mEq/50 mL lots (covering both Exela and Civica brands) include the following lot numbers and expiration dates:
Product was distributed nationwide to wholesalers, distributors, and health systems between January 18, 2022, and February 15, 2023.
Midazolam in 0.8% Sodium Chloride Injection is used for sedation and is packaged in a 100 mL glass vial, 25 vials per corrugated shipper. The vials are labeled with Exela brand (Carton NDC: 51754-2131- 4; Vial NDC: 51754-2131-1, Figure 3).
The affected Midazolam in 0.8% Sodium Chloride Injection 100 mg/ 100 mL include the following lot number and expiration date:
Product was distributed nationwide to wholesalers, distributors, and health systems between July 14, 2023, and September 26, 2023.
ELCYS (cysteine hydrochloride Injection) is used for nutritional requirements per total parenteral nutrition (TPN) and is packaged in a 10 mL glass vial, 10 vials per carton. The vials are labeled with Exela brand (Carton NDC: 51754-1007-3; Vial NDC: 51754-1007-1, Figure 4).
The affected ELCYS (cysteine hydrochloride Injection), USP 50 mg/mL includes the following lot number and expiration date:
The product was distributed nationwide to wholesalers, distributors, health systems, and compounders between July 20, 2023, and August 1, 2023.
Exela is notifying its customers by e-mail and certified mail and is arranging for return and replacement of all recalled product directly to Exela. Customers that have product, which is being recalled should discontinue use, segregate the recalled product, submit a recall stock response form to Exela (even if there is no product to return), and hold the product until shipment instructions are provided by Exela. Customers with questions regarding this recall can contact Exela by phone (828-341-6118) or email at recall@exela.us Monday through Friday, 9:00am – 5:00pm ET. Consumers should contact their physician or healthcare provider if they have experienced any problems related to the usage of this drug product.
(No BeneCard members were affected by this drug recall.)
FDA Reporting Adverse reactions or quality problems experienced with the use of this product may be reported to the FDA’s MedWatch Adverse Event Reporting program either online, by regular mail or by fax. • Complete and submit the report Online: www.fda.gov/medwatch/report.htm • Regular Mail or Fax: Download form www.fda.gov/MedWatch/getforms.htm or call 1-800-332- 1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
Exela Pharma Sciences, LLC, (Exela) is voluntarily recalling the products listed in the table below to the consumer level. Particulate matter identified as silicone was observed during routine inspection of retain samples.
Risk Statement: Administration of an injectable product that contains particulate matter may result in local irritation or swelling in response to the foreign material. If the particulate matter reaches the blood vessels it can travel to various organs and block blood vessels in the heart, lungs or brain which can cause stroke and even lead to death. Exela has not received any reports of adverse events related to this recall.
8.4% Sodium Bicarbonate Injection USP is used for treatment of metabolic acidosis and is packaged in a 50 mL glass single dose vials, 20 vials per carton Exela brand (Carton NDC: 51754-5001-5; Vial NDC: 51754-5001-1, Figure 1) and 25 vials per carton Exela brand (Carton NDC: 51754-5001-4; Vial NDC: 51754-5001-1) and Civica brand (Carton NDC: 72572-740-20; Vial NDC: 72572-740-01, Figure 2). The affected 8.4% Sodium Bicarbonate Injection, USP, 50 mEq/50 mL lots (covering both Exela and Civica brands) include the following lot numbers and expiration dates:
Product was distributed nationwide to wholesalers, distributors, and health systems between January 18, 2022, and February 15, 2023.
FDA Approves Penbraya (meningococcal groups A, B, C, W and Y vaccine) for the Prevention of the Five Most Common Serogroups Causing Meningococcal Disease in Adolescents
Pfizer Inc. has announced that the U.S. Food and Drug Administration (FDA) has approved Penbraya™ (meningococcal groups A, B, C, W and Y vaccine), the first and only pentavalent vaccine that provides coverage against the most common serogroups causing meningococcal disease in adolescents and young adults 10 through 25 years of age. Penbraya combines the components from two meningococcal vaccines, Trumenba® (meningococcal group B vaccine) and Nimenrix® (meningococcal groups A, C, W-135, and Y conjugate vaccine) to help protect against the five most common meningococcal serogroups that cause most invasive meningococcal disease (IMD) globally.
Meningococcal disease is an uncommon but serious illness that can lead to death within 24 hours and, for survivors, can result in life-altering, significant long-term disabilities. Penbraya reduces the total number of doses needed for individuals to be fully vaccinated against the five most common serogroups, thereby streamlining the standard of care, and potentially increasing the number of adolescents and young adults vaccinated.
According to the U.S. Centers for Disease Control and Prevention (CDC), combining vaccines into fewer shots may mean that more adolescents and young adults get their recommended vaccines on time, resulting in fewer delays in protection against serious diseases. Routine use of Penbraya could also reduce IMD cases and associated mortality, the rate of long-term consequences of infection (sequelae) in survivors and costs associated with controlling outbreaks.
FDA Approves Zituvio (sitagliptin) for the Treatment of Adult Patients with Type 2 Diabetes Mellitus Zydus Lifesciences has announced that the U.S. Food and Drug Administration (FDA) approved its New Drug Application (NDA) for Zituvio (Sitagliptin) tablets, 25 mg, 50 mg, and 100 mg. Zituvio contains active ingredient Sitagliptin, a dipeptidyl peptidase-4 (DPP-4) inhibitor indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
Zituvio is a dipeptidyl peptidase-4 (DPP-4) inhibitor indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. Zituvio is not recommended in patients with type 1 diabetes mellitus. Zituvio has not been studied in patients with a history of pancreatitis.
October 19, 2023 – Apotex Corp.; Sun Pharmaceutical Industries, Inc.; Teva Pharmaceuticals USA, Inc., have been approved by the FDA for the use of Pazopanib Hydrochloride Tablets (200 mg/base) for treatment of Renal Cell Carcinoma; Soft Tissue Sarcoma. This approval is as a generic to Votrient.
FDA Approves Wezlana (ustekinumab-auub), an Interchangeable Biosimilar to Stelara
October 31, 2023 — The U.S. Food and Drug Administration has approved Wezlana (ustekinumab-auub) as a biosimilar to and interchangeable with Stelara (ustekinumab) for multiple inflammatory diseases. Wezlana, like Stelara, is approved to treat the following indications:
Adult patients with:
moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy;
active psoriatic arthritis;
moderately to severely active Crohn’s disease; and
moderately to severely active ulcerative colitis.
Pediatric patients 6 years of age and older with:
moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy; and
active psoriatic arthritis.
The FDA’s approval of Wezlana is based on a comprehensive review of scientific evidence demonstrating it is highly similar to Stelara and that there are no clinically meaningful differences between the two products in terms of safety, purity and potency (i.e., safety and effectiveness). This evidence included comparisons of the products on an analytical level using an extensive battery of chemical and biological tests and biological assays that confirmed similarity in the structural and functional features of Wezlana and Stelara (including those known to impact safety and efficacy), and comparative human pharmacokinetic data, clinical immunogenicity data, and other clinical safety and effectiveness data. The evidence also demonstrated that Wezlana met the other legal requirements to be interchangeable with Stelara at the pharmacy level.
Like Stelara, the most serious known side effect of Wezlana is infection. The most common adverse reactions with ustekinumab products are nasopharyngitis, upper respiratory tract infection, headache, fatigue, nausea, vomiting, injection site erythema, vulvovaginal candidiasis/mycotic infection, bronchitis, pruritus, urinary tract infection, sinusitis, abdominal pain, influenza, fever, and diarrhea.
FDA Approves Bimzelx (bimekizumab-bkzx) for the Treatment of Adults with Moderate to Severe Plaque Psoriasis
October 18, 2023 – UCB, a global biopharmaceutical company, has announced that the U.S. Food and Drug Administration (FDA) has approved BIMZELX® (bimekizumab-bkzx) for the treatment of moderate to severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy. Bimekizumab is the first and only approved psoriasis treatment designed to selectively inhibit two key cytokines driving inflammatory processes – interleukin 17A (IL-17A) and interleukin 17F (IL-17F).
The FDA recommended dosage of bimekizumab for psoriasis patients is 320 mg (given as two subcutaneous injections of 160 mg each) at Weeks 0, 4, 8, 12 and 16, then every 8 weeks thereafter. For patients weighing ≥120 kg, a dose of 320 mg every 4 weeks after week 16 may be considered.
Bimekizumab may be administered by a healthcare professional, or a patient may self-inject after proper training. It is available as an autoinjector and a pre filled syringe and will be available in the U.S. in approximately one month.
FDA Approves Velsipity for Moderate-to-Severe Ulcerative Colitis in Adults
The U.S. Food and Drug Administration approved Velsipity (etrasimod) for adults with moderately to severely active ulcerative colitis (UC). Velsipity provides adults living with moderately-to- severely active UC the opportunity to achieve steroid-free remission with an oral, once-daily pill. Approval of Velsipity was granted to Pfizer.
The approval was based on results from the ELEVATE UC phase 3 registrational program (ELEVATE UC 52 and ELEVATE UC 12) that included UC patients who had previously failed or were intolerant to at least one conventional, biologic, or Janus kinase inhibitor therapy. In ELEVATE UC 52, 27.0 percent of patients receiving Velsipity achieved clinical remission versus 7.0 percent of patients receiving placebo at week 12, and at week 52, clinical remission was achieved by 32.0 and 7.0 percent, respectively. In ELEVATE UC 12, 26.0 percent of patients receiving Velsipity achieved clinical remission versus 15.0 percent of patients receiving placebo. At week 12, all key secondary efficacy end points were met, including endoscopic improvement and mucosal healing.
The selective sphingosine-1-phosphate receptor modulator was approved at a 2mg recommended dose. The safety of Velsipity was consistent with previous studies, with the most common adverse reactions being headache, elevated liver tests, and dizziness (incidence ≥5 percent).